

WHEN DO YOU SUSPECT AN INBORN ERROR OF METABOLISM?
If a patient presents with unexplained acute deterioration of their sensorium and clinical status, it is important to suspect an inborn error of metabolism, particularly a small molecule disorder.
Small molecule disorders can occur early in life and can present with symptoms such as poor feeding, repeated unexplained vomiting, seizures, drowsiness, rashes, smells, and tachypnea. Some small molecule disorders are intermittent and manifest during times of stress such as fever, infections, or prolonged fasting.
Large molecule disorders, on the other hand, present with slowly progressive neurological deterioration, which can include coarsening of features, increasing hepatosplenomegaly, progressive loss of intellectual functions, deteriorating vision, increasing seizures, and regression of milestones.
In some lipid storage disorders, accumulation in the eye can present as a cherry red spot. Therefore, early recognition and prompt management of inborn errors of metabolism is critical for improving patient outcomes.
Clues to an inborn error of metabolism:
(Remembered as A, B, C, D......
Acute encephalopathy /Abnormal smell
Bacterial sepsis-like picture
Cardiomyopathy /Consanguinity
Dysmorphism/Death-unexplained/Dermatological
Enlarged liver
Family history
G (jaundice)
Hydrops
Intractable hiccups
COMMON SMALL MOLECULE DISORDER
UCD, MSUD , LACTIC ACIDOSIS , OA
APPROACH TO UREA CYCLE DISORDERS

Drowsiness, poor eating, and vomiting are symptoms of hyperammonemia. After a few days of eating, symptoms typically appear. When there is hyperammonemia without acidosis, a diagnosis is presumed. The problem will be more precisely diagnosed via an amino acid profile.
Protein, benzoate, phenylbutyrate, and oral arginine restrictions are used as treatment.
Except for ornithine transcarbamylase deficiency, which is X-linked recessive, inheritance is autosomal recessive.
MAPLE SYRUP URINE DISEASE (MSUD)

Ketones are detected in the urine, DNPH is detected in the urine, and plasma amino acid levels are raised for branched-chain amino acids. MSUD can be diagnosed if alloisoleucine is found after day 6. Using gas chromatography-mass spectrometry (GC-MS), alpha-hydroxy isovalerate, lactate, pyruvate, and alpha-ketoglutarate can all be found in urine.
Leucine and isoleucine concentrations and the Fisher Ratio (branch-chain amino acids/phenylalanine and tyrosine) are used in tandem mass spectrometry for newborn MSUD screening.
Branch-chain amino acid concentrations in amniotic fluid, mutation analysis, and assessing enzyme activity in cultured amniocytes or chorion villus cells are all examples of prenatal diagnostics.
Dialysis, IV dextrose, insulin, and lipids are all part of acute management. A 4-week trial of thiamine 10-200 mg/day is part of chronic therapy to determine thiamine responsiveness.
Commercial formulations for diets lacking in branched-chain amino acids are available. Although the long-term prognosis is uncertain, some children who underwent liver transplants did well.
ORGANIC ACIDEMIAS

These develop because of a lack of enzymes involved in the breakdown of different amino acids like isoleucine, valine, methionine, threonine, leucine, and lysine, which causes an overabundance of different organic acids. They comprise:
Melanocytic acidemia (MMA)
Propionic acidemia (PA)
Isovaleric acidemia
Glutaric acidemia type I
3-Hydroxy-3-methylglutaryl CoA lyase deficiency
3-methylcrotonyl-CoA carboxylase deficiency
A few days after starting to feed, neonates begin to exhibit symptoms. Lethargy, vomiting, convulsions, rash, strange odors, and a state resembling sepsis appear in them.
The results of the tests may reveal severe lactic acidosis, hyperammonemia, hypoglycemia, thrombocytopenia, and neutropenia.
The abnormal elevations can be seen in the acylcarnitine profile on tandem mass spectroscopy.
Therapy options include eating a low-protein diet, limiting the involved amino acids, taking carnitine and vitamin supplements, and avoiding fasting. The vitamin supplements include biotin in PA and vitamin B12, 1 mg/d in MMA.
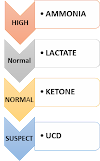
LACTIC ACIDOSIS
1. Check lactic acid is elevated.
2. Check tandem mass spectroscopy.
3. If the acylcarnitine profile on TMS is abnormal consider organic acidemia or fatty acid oxidation defect


5. Use the following algorithm to go further.
4. If TMS is normal check pyruvate, blood sugar and cardiac and multisystem involvement.
PDH (PYRUVATE DEHYDROGENASE )
PC (PYRUVATE CARBOXYLASE)
TRYING TO MAKE THINGS EASIER.
Dr.Amit kumar


0 Comments